If we know how a specific amino acid sequence folds, then researchers can uncover what the protein does; we can start to understand and anticipate the protein’s properties and function. The issue, however, is that there are 10¹⁹⁸ ways for any sequence to fold (there are ~10⁸⁰ atoms in the observable universe). Levinthal’s paradox revolves around the fact that if a protein were to sample every possible ternary structure sequentially, it would take longer than the age of the universe to arrive at its correct conformation, even if a permutation was tried every picosecond. The paradox lies in the fact that most proteins fold spontaneously on a milli- or even microsecond time scale.
There are an estimated 200 million distinct proteins, of which we only know the 3D structure of about 170,000. The current methods of deciphering the tertiary structure are expensive, lengthy, and require a significant amount of trial-and-error. The most notable one is X-ray crystallography, which operates by firing incident rays and measuring their angle and intensity diffraction from the crystalline structure takes a year to complete and costs ~120,000 USD. Other methods include nuclear magnetic resonance and cryo-electron microscopy. These methods are too costly and have too much inherent uncertainty. Alphafold changes everything, fundamentally solving the protein folding problem: predicting how protein chains will fold given an arbitrary sequence of amino acids. The protein folding problem has been a grand challenge in biology for 50 years. And it’s been solved decades before many researchers antipiated.
This has been a problem plaguing scientists and researchers for decades, with the advancements resulting from the marriage between biology and engineering. There has been a myriad of developments, from IBM’s Blue Gene spearheading supercomputing efforts to efforts from the scientific population, most notably Folding@Home and FoldIt. The key to the solution, however, lies in the field of deep learning.
Biologists are turning to AI methods as an alternative, with the ability to computationally analyze an amino acid sequence and generate a prediction for the structure of that protein accelerating research and allowing for extensive scalability. Thanks to the immense amount of data available, AI has blossomed in a non-traditional area.
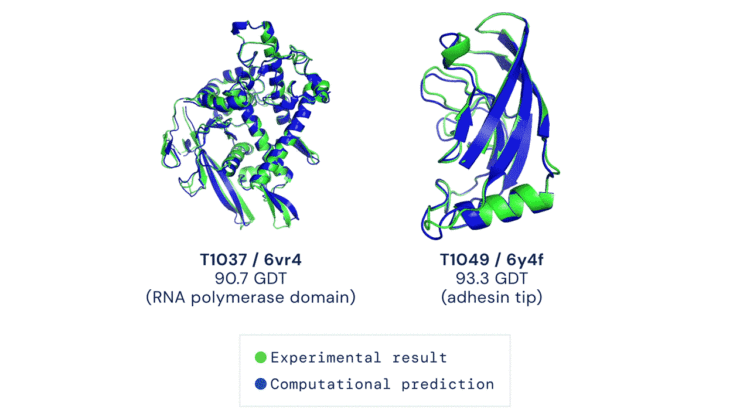
Progress on the protein folding problem is measured at a biennial global competition, CASP (Critical Assessment of protein Structure Prediction). CASP is the golden standard for solutions, with everyone from academics to billion-dollar companies submitting solutions. This year (CASP 13), DeepMind’s AlphaFold placed first at the competition, with a global distance test (GDT) score, a measure of how similar the predicted structure is to the actual structure, of 92.5 i.e. an average error of 0.1 nanometers. For comparison, 90 is the equivalent for experimental methods. AlphaFold, equipped with algorithms and data, produced one of the most extraordinary results in structural biology and genomics.